成功CRISPRi的5个关键步骤

基因组编辑技术正在彻底改变生命科学,而CRISPR技术是定期更新其多功能平台的关键参与者。在选定的细菌和古生菌中,CRISPR (C光泽R鲁西平原我nterspaced年代长的矮PalindromicRepeats)和CRISPR相关系统(Cas)通过碱基对形成rna靶向并切割外源DNA元件,以授予适应性免疫。该机制在实验室中实验使用(和优化)在宿主独立的环境中,以目标和编辑特定的遗传密码或重编程基因组来操纵细胞。虽然不同的CRISPR系统存在于各种生物中,但最简单的版本是II型CRISPR,由一个Cas9催化酶和两个rna组成;成熟的CRISPR RNA (crRNA)和反式作用RNA (tracrRNA)。研究人员结合双组分原生RNA系统,在实验室中设计了嵌合单导RNA (sgRNA),提高了实验效果。2012年,一个科学家团队通过整合核酸酶缺陷、突变的Cas9或“死亡的Cas9”(dCas9),对CRISPR系统进行了进一步的变异,以重新利用该系统,并在不切割/编辑的情况下调节DNA序列进行改变和抑制,从而产生CRISPR干扰(CRISPRi)。
CRISPRi同化催化死亡的Cas9,缺乏内切酶活性,与引导RNA (gRNA)共表达。然后dCas9:sgRNA机制形成DNA识别复合体,该复合体可以特异性地干扰转录延伸或与RNA聚合酶(RNAP)位点结合以阻止转录启动,从而全面阻断信使RNA (mRNAs)的产生。RNA嵌合体和目标DNA之间的碱基对形成由位于目标序列3 '端的短DNA序列实现,称为“原间隔相邻基序”(PAM)(图1)。失活的dCas9突变体是通过ruvc样(D10A)和HNH核酸酶(H840A)结构域的两个点突变实现的。dCas9:sgRNA机制可以与额外的效应域融合,用于有效的基因干扰(CRISPRi)或基因激活(CRISPRa)。
CRISPRi的主要亮点(包括优点和缺点)
- 该机制由一个非活性的dCas9蛋白定义,用于靶向基因组调控。
- dCas9: sgRNA机制允许通过sgRNA的20个碱基对区域形成互补碱基对,以靶向感兴趣的DNA序列上的特定基因组位点。
- CRISPRi可以通过多个rna调控多个基因而不产生脱靶效应。
- 这种沉默是可诱导和可逆的,在细菌中具有高度特异性。
- 在哺乳动物细胞中,转录抑制是相对可变和适度的。
- 相关外源DNA上必需的PAM序列可能限制结合位点。
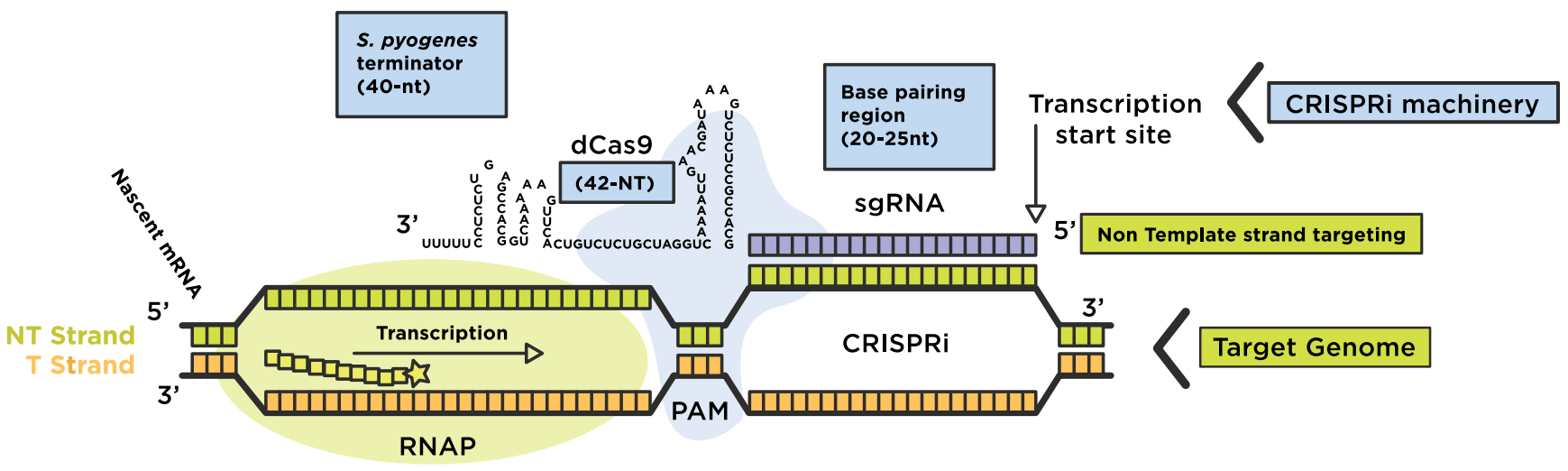
图1:重新利用CRISPRi系统来调节基因组。dCas9-sgRNA复合物与目标DNA区域的非模板链形成碱基对,用于转录干扰(延伸或起始)。原图片改编自Addgene.资料来源:Lei S Qi(斯坦福大学)
CRISPRi机制提供了一种高效、可逆和特异性的基因组调控平台,用于转录控制,而不改变目标DNA序列。以下指南包括使用派生的dCas9实现CRISPRi的关键步骤酿脓链球菌.
Would你更喜欢以PDF格式阅读?
在这里下载
计划CRISPRi实验-实验设计
一旦你决定了所需的基因操作,使用以下变量来设计哺乳动物细胞和细菌生物中的CRISPRi实验框架。
- 选择Cas9的种,设计死Cas9 (dCas9)。
- 设计sgRNA序列用于特定位点的碱基配对,使脱靶效应最小化。
- 选择Cas9和sgRNA的表达系统。
- 选择一个可选择的标记物(要量化的药物或荧光团)
- 选择交付方式。
- 选择检测方式。
选择CRISPRi的目标位点
dCas9-sgRNA复合物与目标DNA的结合特异性依赖于NGG PAM基序。对于基因组内灵活的靶点特异性选择,CRISPRi靶向是通过sgRNA互补的20碱基对(bp)区域和目标DNA序列之间直接的Watson Crick碱基配对,通过必需的3-nt PAM序列实现。
- 其他来自不同物种的Cas蛋白及其对应的PAM序列可在Addgene.org上获得。
- 嵌合sgRNA包含一个20个核苷酸(nt)碱基配对区域2)一个dCas9处理发夹(42 nt)和3)一个链球菌衍生终结者(40-nt)来模拟天然RNA复合物。
- 设计20-nt sgRNA序列,以目标基因的启动子/增强子区域为目标,或以目标基因5 '端编码序列的开头为目标。
- 使用Basic Local Alignment Search Tool (BLAST)检查sgRNA在基因组中结合的特异性。为了排除额外的结合位点和脱靶效应,BLAST在感兴趣的基因组中搜索14-nt特异性区域,其中包括sgRNA的12-nt“种子区域”和3-nt PAM区域的2-nt。
- 对于大量短核苷酸sgrna的基因组图谱,使用SeqMap计算脱靶效应。只使用没有可预测脱靶的sgrna。
- 附加dCas9句柄和链球菌利用Quickfold或RNAfold模拟算法预测嵌合sgRNA二级结构。
- 确认sgRNA序列不包含限制性内切酶位点序列(例如细菌sgRNA的EcoRI、Bg1II和BamHI;BstXI和XhoI(用于人类sgRNAs)),然后将模板克隆到表达载体中。
- 可选步骤:调节抑制效率,在sgRNA碱基配对区域引入单个或多个错配。
选择Cas9和sgRNA的表达系统
在哺乳动物细胞中,催化死亡的Cas9 (dCas9)可以融合到转录抑制因子域,如KRAB (Krüppel associated box),以增强抑制作用。相反,激活基因表达使用激活域,如VP16或VP64,与哺乳动物细胞中的dCas9配对。为了使CRISPRi功能化,你需要通过表达载体在目标细胞中表达的dCas9和gRNA模板。
- 为sgRNA和dCas9选择合适的表达载体。在哺乳动物细胞中使用的主要表达系统和变体是网上.
- 用于哺乳动物系统和细菌中的CRISPRi质粒或载体表达系统浏览可用的结构.
- 以下由Stanley Qi实验室重新放置的质粒,推荐用于细菌生物和哺乳动物细胞中dCas9和sgRNA的表达。
- 如果你使用的质粒不共表达gRNA和dCas9,使用单独的表达载体来抑制特定的感兴趣位点,如上所述。
- 当克隆嵌合sgRNAs到一个小表达载体(5 kb)时,使用寡聚PCR和消化/连接方法,以避免iPCR可能引入的PCR错误。
- 要将多个sgrna克隆到单个表达向量,请使用倡导装配方法或金门克隆程序复用CRISPRi。
- 这些载体可以转化为大肠杆菌的克隆菌株,例如市售的One Shot Top10化学活性菌株大肠杆菌细胞,然后培养到足够数量的质粒纯化。
- 表达系统应包括选择标记(如新霉素)或报告基因(如GFP),以在传递到靶细胞后验证基因修饰。
- sgRNA表达的阴性对照是一个没有20-nt碱基配对区域的表达载体。dCas9表达的阴性对照是相同的表达向量,但没有dCas9编码序列。
下发dCas9和sgRNA
我使用来自LI-COR的IRDye偶联抗体,可以通过他们的奥德赛®成像系统检测。这些染料可以在700nm或800nm通道中检测到。
- 对于细菌系统中的靶向基因表达:将sgRNA载体与诱导的dCas9表达载体(例如#44249)共同转化为所需的sgRNA载体大肠杆菌菌株(如MG1655)。
- 对于哺乳动物细胞(例如HEK 293细胞-人胚胎肾细胞系)中的靶向基因抑制,在转染前将细胞系在补充的生长培养基中培养。
- 使用市售的DNA转染试剂转染哺乳动物细胞,遵循制造商的协议。
- 每个培养细胞均转染良好的dCas9表达质粒和sgRNA表达质粒。
- 对于哺乳动物细胞,在转染72小时后测量基因表达。
设计功能验证分析
一旦gRNA和dCas9成功地传递到目标细胞,就是时候验证预期的CRISPRi调控或基因组抑制水平了。
- 如果目标基因与荧光蛋白或LacZ融合;使用流式细胞仪或β-半乳糖苷酶活性测定作为功能测定来测量对蛋白质表达的影响。
- 测量内源性基因转录抑制的功能测定也可以包括qRT-PCR或全基因组NET-Seq技术。
- 例如,Net-Seq可以通过dCas9: sgRNA机制的干扰指示被破坏的确切基因组位点。
- 由于CRISPRi的功效可能因细胞类型和生长条件而异,因此在相同细胞类型和相似生长条件下进行所有测试。
- 这里列出的概述指南描述了CRISPRi (sgRNA和dCas9表达抑制感兴趣的靶基因)在大肠杆菌(如MG1655株)或人类细胞(如HEK 293细胞)。
- 在之前的研究中,dCas9和sgrna在细菌中促进了强大的沉默和基因抑制,效率更高(99-99.5%),而在人类细胞中效率中等(~50-60%),并有可能进行优化。
参考文献
- 拉森先生;吉尔伯特·l·;王x;Lim w;斯曼j .;CRISPR干扰(CRISPRi)用于基因表达的序列特异性控制。Nat Protoc. 2013;8(11), 2196。
- Weiyue j .;et al。通过细菌偶联转移的CRISPRi系统特异性基因抑制。美国化学学会,合成生物学,2014;3(12), 931。
- 气,l;拉森先生;吉尔伯特·l·;Doudna j .;斯曼j .;阿金a;Lim w;重新利用CRISPR作为rna引导的基因表达序列特异性控制平台。细胞。2013;152年,1183年。
- Jinek m;et al。可编程双rna引导的DNA内切酶在适应性细菌免疫。科学。2012;337年,821年。
- 井上h;Nojima h;日本冈山h;用质粒高效转化大肠杆菌。基因1990;96:23-28。
- 罗伊·k;史密斯j .;Vonesch美国;林g;你c;莱德尔a;楚a;苏雷什美国;阮M;特里帕西a; et al, Multiplexed precision genome editing with trackable genomic barcode in yeast. Nature Biotechnology, 2018; 1696
- Charpentier E.和Doudna J.重写基因组。《自然》、《新闻与观点》2013;495年,51
- 跑f;许p;赖特j .;《诉;斯科特·d·;张峰。利用CRISPR-Cas9系统进行基因组工程。《自然议定书》,2013;8 (11) 2281
- 迈尔斯美国;赖特j .;Peckner r;卡利什b;张f;通过dcas9 - apex介导的接近标记发现与预先定义的基因组位点相关的蛋白质。Nature Methods, Brief Communication, 2018;5.
- Hannon, g.j., RNA干扰。自然,2002;418年,251年。
- Beerli, r.r.,和Barbas, c.f.,工程多指锌指转录因子。生物技术,2002;20日,141年。
- 王慧华,艾萨克斯,冯杰,卡尔,孙志忠,徐国强,林志荣,丘奇,孙志强。多重基因组工程与加速进化的细胞编程。自然,2009;460年,898年。
- Ledford H.,驾驭CRISPR浪潮,自然,新闻专题,2016;531年,159年


